
Introducción
Muchas muestras para cromatografía de gases (GC) contienen cantidades significativas de compuestos no volátiles en la matriz de la muestra. La inyección directa causa que solutos fuertemente retenidos o materiales no volátiles residuales se mantengan en el sistema cromatográfico y puedan acumularse hasta causar interferencias con los analitos de interés.
Los síntomas típicos son menores áreas, picos con cola, la formación de productos de degradación volátiles, sangrado de columna y un mayor número y tamaño de picos fantasma. La introducción de una gran cantidad de compuestos extraños puede comprometer las prestaciones del instrumento mismo que obligan a la substitución del inserto del inyector, la eliminación de parte de la columna, de la precolumna si está instalada y, eventualmente, el cambio de la columna analítica.
Si en la muestra hay compuestos relativamente poco volátiles pero cromatografiables, se puede usar la técnica del “backflushing” que, mediante la inversión del flujo de la columna permite la eliminación de los compuestos menos volátiles.
Otros métodos de preparación de la muestra permiten inyecciones más limpias mediante derivatización, extracción o filtrado que separan o eliminan los compuestos conflictivos, pero a costa de una mayor manipulación de la muestra a veces con reactivos tóxicos o peligrosos, y menores recuperaciones y reproducibilidades.
El muestreo por HS evita que los residuos no volátiles de la muestra entren en el Cromatógrafo de Gases manteniéndolos en un vial y transfiriendo los componentes volátiles al inyector y columna.
Los cromatografistas generalmente dividen la técnica en dos clases, HS Estático y HS Dinámico, que hacen referencia al método de extracción de los compuestos volátiles de la muestra. En HS Estático los analitos pasan a la fase gas mediante procedimientos térmicos y químicos mientras que en HS Dinámico los analitos se eliminan dinámicamente por purga mediante un gas inerte.
Ambas técnicas son eficaces y versátiles, pues la mayoría de las muestras pueden analizarse con cualquiera de las dos técnicas y la elección de una u otra depende de las regulaciones existentes. Por ejemplo el análisis de VOC’s en intermedios farmacéuticos se efectúa por HS Estático según United States Pharmacopeia National Formulary o métodos similares Europeos, mientras que la determinación de VOC’s en agua potable se efectúa por HS Dinámico según USEPA Method 524.2.
HS Dinámico
En HS Dinámico la muestra se mantiene en un recipiente por el que se hace pasar un gas inerte, generalmente gas portador, durante el período de muestreo. Los VOC’s purgados de la muestra se retienen en una trampa de adsorción o se condensan en una trampa fría, de donde posteriormente se desorben térmicamente y pasan a la columna.

El HS Dinámico es la base de los análisis por Purga y Trampa (Purge&Trap) de VOC’s en agua.
Con una sistema dinámico Se obtiene una mayor sensibilidad, ofrece hasta 100X de incremento en sensibilidad frente

a las técnicas convencionales de Espacio de Cabeza, mediante la purga constante de los compuestos volátiles de la muestra termostatizada y su enriquecimiento en la trampa adsorbente.
Mediante el uso le la combinación adecuada de temperatura y naturaleza del adsorbente, se consigue la concentración eficiente de analitos a nivel de trazas en un amplio rango de volatilidad.
Algunos ejemplos de fabricantes de este tipo de unidades son Tekmar Aquatek 100, o Dani Master DHS (Ver Figura 2).
HS Estático
En este caso la muestra se mantienen sellada en un vial hermético
(de 6, 10 o 20 mL) cerrado mediante cápsula o tapón y una membrana adecuada que, generalmente, es de silicona con una lámina de PTFE. El vial y la muestra se mantienen en condiciones de temperatura controlada para permitir que los compuestos volátiles (VOC’s) pasen de la fase sólida a la fase gas del vial. La zona gas del vial que, obviamente, no se llena del todo con la muestra, es lo que se denomina Espacio de Cabeza. Una vez que una parte de la fracción volátil pasa a la fase gas, se transfiere una parte de la misma a la columna cromatográfica.
En HS Estático hay que permitir una combinación de tiempo de incubación y temperatura para que la concentración de los analitos volátiles se mantenga estable y alcance un equilibrio antes de la extracción y transferencia. En muestras sólidas o polímeros resulta difícil alcanzar un estado de equilibrio y se efectúan etapas de extracción y análisis múltiples, uno por etapa, o se acumulan los productos extraídos en una trampa seguida de desorción y un análisis único.
El Equilibro en HS
Al equilibrio el sistema químico de la muestra en el vial afecta directamente a la transferencia de VOc’s hacia la columna.

La Figura 3 muestra un sistema típico con sus características físico-químicas más importantes como el Volumen de la Fase Líquida VS y de la fase gaseosa VG, la Concentración de los analitos tanto en Fase Líquida CS como en Fase GAS CG, y la migración de las moléculas de soluto entre las dos fases.
La concentración en la fase gas de cada analito es la cantidad medida en el análisis por GC, no sus concentraciones en la fase líquida. Para determinar las concentraciones originales del analito C0, hay que considerar el coeficiente de reparto K en condiciones de equilibrio. K es la relación de las concentraciones de un compuesto en la fase líquida y en la gaseosa:

El coeficiente de distribución depende de la temperatura y de la naturaleza química de la fase líquida en relación con los solutos. Normalmente K disminuye al aumentar la temperatura, es decir un aumento de las concentraciones en la fase gas, y aumenta con composiciones de la fase líquida en la que los analitos son más solubles.
La relación de los volúmenes de la fase gas frente a la fase líquida, β, tiene un papel secundario pero muy importante en la determinación de las concentraciones de analito en las dos fases al equilibrio:

Se puede demostrar que la concentración original del soluto en la fase líquida está relacionada con su concentración en la fase gas, el coeficiente de distribución y la relación de fases por esta ecuación, denominada a veces como Ecuación de Espacio de Cabeza:

Notar que la suma de K y β controla las concentraciones en fase gas y líquida, no separadamente.
En HS Estático, resulta imprescindible el control de K y β, mediante un estricto control de la temperatura, del volumen y preparación de la muestra, para mantener esencialmente constantes los dos parámetros a lo largo de una serie de análisis y efectuar una fácil calibración mediante métodos de Adición Estándar o Estándar Externo. K y β acaban siendo parte del coeficiente de calibración instrumental para cada analito además de otros factores como el volumen de gas transferido a la columna y el factor de respuesta del detector.
La concentración de equilibrio en la fase gas del espacio de cabeza, medida por el GC, puede derivarse de la ecuación anterior así:
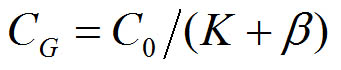
Cuando la suma de K y β disminuye, la concentración en la fase gas aumenta. K es función de la temperatura y de las características químicas del sistema cerrado del vial, y β es función de los volúmenes de muestra y gas en el vial.
Veamos como diferentes situaciones de esos factores afectan tanto el tamaño de los picos y su repetibilidad para un analito muy soluble en agua, etanol, y otro mucho menos soluble como el hexano.
Efecto de la Temperatura
El coeficiente de distribución de ambos analitos en agua depende de la temperatura de manera logarítmica. La Figura 4 muestra el coeficiente de distribución del etanol en agua en función de la temperatura, con un valor entre 200 y 1350 en el rango de temperaturas entre 80-40ºC.
El n-hexano es relativamente insoluble en agua y su coeficiente de distribución cambia con la temperatura en una magnitud relativa similar al etanol, pero los valores absolutos varían entre 0.01 y 0.15 en el mismo rango de temperatura.

Para compuestos de alta solubilidad en la fase líquida como el etanol en agua, donde K>>β, el coeficiente de distribución controla la concentración en la fase gas según la Ecuación 4, de tal manera que pequeños cambios en la temperatura del vial causan grandes cambios en la concentración en la fase gas y en el área de los picos.
La Figura 5 muestra los cambios en el área relativa de los picos al aumentar la temperatura del sistema etanol-agua para volúmenes constantes de fase líquida en el vial, con incrementos de 6.3X pasando de 40 a 80ºC.

Para compuestos muy solubles como el etanol las áreas de los picos son muy sensibles a pequeños cambios de temperatura: un incremento de 60 a 62ºC incrementará el área del etanol en casi un 10%.
Inversamente para compuestos de baja solubilidad como el n-hexano en agua donde K<<β, la influencia del coeficiente de distribución es pequeña y los cambios de temperatura tienen poco efecto, con un factor de incremento de las áreas de 1.3X pasando de 40 a 80ºC. Un cambio de 2ºC, produce efectos negligibles de aproximadamente 0.1%.
Efecto del Volumen de Muestra
El volumen de la fase líquida, ver Ecuación 2, tiene una fuerte influencia en la concentración en la fase gas si el compuesto es poco soluble. En el caso del n-hexano, donde K ≈ 0.04 a 60 °C, resulta que β >> K en un amplio rango de volúmenes de muestra, por lo tanto cambios en el volumen de la muestra afectarán fuertemente la concentración del soluto en la fase gas. La Figura 6 muestra un incremento relativo de 6.8X al pasar de un volumen de muestra de 5 a 15mL a una temperatura constante de 60ºC. Un incremento de 0.2 mL causará un aumento de las áreas de un 5%.

Para el sistema etanol/agua, donde K ≈ 500 a 60 °C, los cambios en la comparativamente pequeña relación de fase no tienen prácticamente influencia en la concentración de la fase gas. La Figura 6 muestra una línea prácticamente recta de las áreas del etanol entre 5 y 15mL de volumen de muestra, donde el área aumenta únicamente un 0.5%. El efecto de un incremento de 0.2mL es despreciable.
Como obtener mayor sensibilidad
Algunas muestra no permiten el suficiente límite inferior de cuantificación (LOQ) deseado. Como en un sistema de Espacio de Cabeza la sensibilidad depende tanto de las solubilidades como de las relaciones de fase de los analitos: la sensibilidad puede aumentarse reduciendo tanto K como β.
En técnicas de inyección directas, muestra mayores producen picos más grandes para todos los compuestos en proporcione iguales, y puede parecer que en HS valga la misma regla, pero de hecho este efecto es parcialmente cierto.
Si se consideran las áreas relativas del etanol y n-hexano de la Figura 6, los datos son para volúmenes mayores de la misma concentración en agua de cada componente, aunque sólo el n-hexano muestra áreas que incrementan con el volumen de muestra.
Hay poca ganancia al aumentar el volumen de muestra con analitos muy solubles como el etanol en agua. Con una amplia relación de reparto, las menores relaciones de fase que resultan de mayores volúmenes de muestra no influenciarán la suma de K y β, y, por lo tanto, las áreas no incrementarán.
Hay que tener en cuenta que los muestreadotes de HS tienen un límite en el volumen de la muestra impuesto por la posición más inferior de de la aguja de muestreo, ya que resulta imperativo evitar cualquier posibilidad de que haya contacto con la fase líquida que contaminará la aguja y causará contaminaciones cruzadas.
Una mayor temperatura de termostatización tiene una fuerte influencia en la constante de distribución y pueden usarse como ventaja, con un límite. Para compuestos solubles como el etanol en agua, una mayor temperatura causará mayores sensibilidades pero no es el caso con compuestos poco solubles como el h-hexano, que produce áreas poco dependientes de la temperatura.
Se presenta otro problema: la presión de vapor del agua aumenta exponencialmente con la temperatura, así como otros solventes comunes. Si se usa una temperatura muy alta del vial, se puede producir una explosión del vial (excepto si usa cápsulas con válvula de sobrepresión), además algunas muestras pueden empezar a descomponerse a mayores temperaturas con resultados analíticos peores.
Como alternativa al aumento de temperatura de muestras HS acuosas, la adición de una sal como NaCl o K2CO3 desplazará la distribución de los compuestos muy solubles hacia la fase gas. En general hace falta una gran cantidad de sal para obtener un efecto significativo en muestras acuosas, del orden de 20-50% w/v.
La sal puede ser una fuente de interferencia si no se trata a alta temperatura y no se mantiene en contenedores convenientemente sellados para evitar la adsorción de VOC’s del aire ambiente.
Como optimizar el método
Puede que amplios cambios en el volumen de muestra y en la temperatura de termostatización no estén dentro de los límites de situaciones reguladas. Aún en esas situaciones, es posible aumentar la calidad de los resultados si se presta más atención a la técnica y en la condición y calibración del instrumento.
Como se ha indicado las áreas de los picos en análisis por HS dependen de la temperatura y el volumen de muestra. Mientras que los efectos de la temperatura dominan los compuestos altamente solubles, los efectos del volumen dominan a los menos solubles. Los analitos con solubilidades intermedias pueden experimentar un efecto mixto tanto del volumen como de la temperatura.
Inestabilidades en la temperatura de sólo 2ºC pueden causar un variación de un 10% en el área de los picos como el etanol, mientras que una incertidumbre volumétrica del 0.2 mL producirán un 5% de variación para compuestos como el n-hexano. Es, por lo tanto, muy importante que tanto el volumen del vial como de la muestra líquida sea lo más precisa y repetitiva en aras de obtener la máxima repetibilidad en los resultados.
La precisión, estabilidad y consistencia de la temperatura de termostatización resulta también crítica. Los muestreadotes modernos por HS tienen una excelente regulación, pero alanos sistemas más antiguos pueden ser deficientes en este aspecto. El fabricante deberá proveer del procedimiento adecuado para la comprobación y validación de la variabilidad de la temperatura vial a vial, que puede efectuarse con un solución acuosa de etanol al 0.1% v/v.
El uso del tipo de vial adecuado también es extremadamente importante respecto a la temperatura. Cada fabricante deberá especificar los tipos de vial certificados para HS, pues los que ajustan mal no sólo tendrán temperaturas de incubación variables sino que además no se calentarán a la misma velocidad, con lo que resultará afectado el tiempo de termostatización. Viales no adecuados pueden romperse en la zona calefactada, con la consiguiente pérdida de operatividad durante la resolución de la incidencia.
Instrumentación
Las tres técnicas más usuales utilizadas para la transferencia una muestra de espacio de cabeza estático son:
- Jeringa para gases
- Compensación de presión
- Bucle a presión
Inyección con Jeringa para Gases (Gas-Tight Syringe Injection)
Esta técnica opera termostatizando inicialmente la muestra en un horno de incubación a una temperatura y tiempo determinados hasta que se alcance un estado de equilibrio (ver Figura 7, Etapa 1). Una vez alcanzado el equilibrio, una alícuota se toma del espacio de cabeza con una jeringa hermética para gases (Figura 7, Etapa 2) y se inyecta en el GC como si fuera una muestra líquida (Figura 7, Etapa 3).

Hay que tener en cuenta los siguientes consideraciones con esta técnica.

Puesto que la muestra se transfiere de un horno caliente, la jeringa también ha de mantenerse caliente para asegurar que la muestra no vuelva a condensarse en la jeringa. Muchos fabricantes tienen en cuenta este punto y sus inyectores disponen de un horno de calefacción para la jeringa.
Hay posibles problemas de reproducibilidad debido a posibles pérdidas de muestra. Puesto que la muestra se transfiere de un vial al inyector, parte puede perderse debido a diferencias de presión entre el vial y las condiciones ambientes.
Más allá de estas consideraciones, la técnica con jeringa es simple, se puede aplicar a una gran variedad de equipos, y resulta más adecuada para muestras diversas.
Algunos ejemplos de fabricantes de este tipo de unidades son ThermoQuest TRACE™ HS2000 y HS850 y Leap Technologies CTC COMBI (ver Figura 8).
Inyección con Compensación de Presión
Otra técnica usual es el sistema con compensación de presión, que permite obtener resultados con un elevado grado de repetibilidad.

Se utiliza una inyección directa desde el vial al gas portador sin otras partes móviles más que una válvula y una aguja. Este sistema, como otras técnicas, usa un horno de incubación para termostatizar el vial hasta que la muestra alcanza el equilibrio (Figura 9, Etapa 1).

Durante estas etapas iniciales, se introduce en el vial una aguja que se presuriza con gas portador (Figura 9, Etapa 2). Tras la pesurización del vial y llegar al equilibrio, la válvula se conmuta durante un tiempo determinado para que la muestra pase a la línea de transferencia y a la columna (Figura 9, Etapa 3).
Puesto que esta técnica usa un período de tiempo teórico para inyectar la muestra, el volumen absoluto de la misma no se conoce. Sin embargo esta técnica resulta altamente reproducible puesto que el número de partes móviles se minimiza, evitándose el riesgo de adsorciones y pérdidas debido a fugas. Un ejemplo de un sistema de este tipo es el TurboMatrix HS-16 fabricado por Perkin-Elmer (Figura 10).
Inyección con Bucle a Presión
La última técnica usual es la Inyección con Bucle a Presión.

A diferencia del sistema con compensación de presión, el sistema con bucle a presión usa una cantidad de muestra conocida. Esta técnica usa generalmente un válvula de seis vías, e inicialmente termostatiza y presuriza el vial como en la técnica anterior (Figura 11, Etapa 1). Tras la presurización, se conmuta la válvula y el bucle se llena con la muestra (Figura 11, Etapa 2). E la última etapa la válvula se conmuta de nuevo para redirigir el flujo de portador para pasar la muestra contenida en el bucle a la línea de transferencia y a la columna (Figura 11, Etapa 3).

Esta técnica tiene algunas ventajas y desventajas. Una de las ventajas es que el bucle puede termostatizarse a altas temperaturas para limitar la adsorción de compuestos de alto peso molecular y sensibles.. El volumen fijo del bucle permite mejorar la reproducibilidad entre determinaciones. Una desventaja de la técnica es la posible contaminación cruzada con muestras anteriores que pueden producir picos fantasma. Hay varios fabricantes de este tipo de inyectores como Tekmar 7000HT, HP 7694E y DANI MASTER SHS (Ver figura 12).
Consideraciones sobre la Microextracción en Fase Sólida (SPME) en HS
La Microextracción en Fase Sólida (SPME) es una técnica de extracción sin solvente que se basa en la exposición de una fibra de sílice fundida recubierta con una fase estacionaria al Espacio de Cabeza hasta que se alcanza un equilibrio entre los analitos volátiles en la fase gas y en la fibra. El analito se resorbe de la fibra a alta temperatura en un inyector cromatográfico, generalmente un GC, donde se analiza.
El concepto es similar al de Purge&Trap pero es más simple y puede implementarse en un inyector automático que permita todos los sitema de inyección.
Principios básicos de la SPME
SPME se basa en el equilibrio de reparto de los analitos objetivo entre una fase polimérica depositada sobre una fibra de sílice fundida y la matriz de la muestra. Para la extracción en SPME no se requieren solventes orgánicos.
La masa extraída y el rango lineal dependen de los coeficientes de reparto (KfS) y el volumen de la fase estacionaria.
La transferencia de los analitos desde la matriz al recubrimiento si inicia en el momento que la fibra recubierta se pone en contacto con la muestra. Las condiciones de equilibrio pueden describirse como:

donde n es la cantidad extraída por el recubrimiento, KfS. Es la contante de distribución recubrimiento/matriz, Vf es el volumen de recubrimiento de la fibra, VS es el volumen de la muestra, y C0 es la concentración inicial de un analito en la muestra.
La ecuación muestra que hay una relación proporcional entre la concentración de la muestra y la cantidad de analito extraído que permite su cuantificación.
Modos de Extracción
En SPME se pueden efectuar dos tipos básicos de extracción: la Extracción Directa y la extracción por Espacio de Cabeza (HS).

En el primer caso la fibra se introduce directamente en la muestra acuosa o gaseosa y los analitos se transfieren directamente desde la

matriz de la muestra a la fase de extracción. Con muestras gaseosas la convección natural es suficiente para facilitar un equilibrio rápido, pero con muestras acuosas es necesario un cierto nivel de agitación.
En el modo espacio de cabeza los analitos han de transportarse a través de la barrera del aire antes de que alcance el recubrimiento. En este caso se evita el daño causado a la fibra por parte de interferencias no volátiles de alto peso molecular que puedan estar presentes en la matriz. El modo HS permite también modificar la matriz (cambio de pH, variación de la fuerza iónica) sin dañar la fibra.
Si los volúmenes de la muestra y del espacio de cabeza son los mismos en cada determinación, las cantidades de analito adsorbidos en la fibra en el equilibrio se mantienen constantes.
Parámetros experimentales que afectan la eficacia de extracción
La cantidad de analito extraído depende del reparto entre la matriz (acuosa en SPME directa o gaseosa en SPME HS) y la fibra que depende por la constante de distribución. Puesto que la cantidad de analito extraído determina la sensibilidad de método la elección de los parámetros óptimos (tanto en la etapa de extracción como la de desorción) que controlan la SPME resultan de gran importancia. Los principales son:
Etapa de Extracción
- Elección del recubrimiento de la fibra
- Tiempo de extracción
- Temperatura
- Agitación
- Fuerza iónica
- pH
- Volumen de muestra
- Efectos matriz
- Posición de la fibra
Etapa de desorción
- Tiempo de desorción
- Temperatura del Inyector
Un equipo que permite esta técnica es Leap Technologies CTC COMBI (ver Figura 14).
Cómo contactar con Cromlab
Para cualquier información adicional u oferta de productos contacte con nuestro Departamento Comercial.